A density functional theory study of catalytic sites for oxygen reduction in Fe/N/C catalysts used i
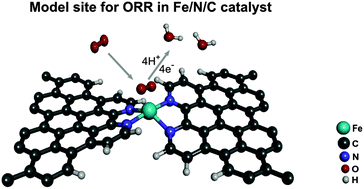
The oxygen reduction catalytic activity of carbon-supported FeN4 moieties bridging micropores between two graphene sheets was investigated by density functional theory (DFT). Based on the FeN2+2/C structure proposed earlier by our group, two types of FeN2+2/C structures were considered: one mostly planar and one in which the Fe ion is significantly displaced out of the graphitic plane. A structure in which the FeN4 moiety is embedded in an extended graphene sheet (FeNpyri4/C) was also considered. In addition, we have investigated the influence of an axial pyridine group approaching the Fe centre. The formation energy is lowest for the planar FeN2+2/C structure. The overall downhill behaviour of the relative free energy vs. the reaction step suggests that most structures have catalytic activity near zero potential. This conclusion is further supported by calculations of the binding energies of adsorbed O2 and H2O and of the O–O bond lengths of adsorbed O2 and OOH. The side-on interaction of adsorbed O2 is preferred over the end-on interaction for the three basic structures without the axial pyridine. The pyridine coordination produces a stronger binding of O2 for the planar FeN2+2/C and the FeNpyri4/C structures as well as a dominant end-on interaction of O2. The energy levels of the planar FeN2+2/C structure with and without the pyridine ligand are nearly equal for iron spin states S = 1 and S = 2, suggesting that both configurations are formed with similar concentration during the preparation process, as also previously found for two of the iron sites by Mössbauer spectroscopy experiments.